Overview
Firstly, I’d like to briefly explain our study to give some background to the analysis I carried out (more detail can be found in the paper: link here when accepted). Our study aimed to characterise the microbiome of the nasopharynx (the area at the back of the nose) and middle ear in children with recurrent ear infections. We also compared the nasopharynx of these children to that of healthy children to see if we could find commensal (“good”) bacteria that might protect against recurrent ear infections. We ended up with almost 100 children in each group and had over 450 samples in total for analysis.
Prior to sequencing
As this document focuses on the data analysis, I will not go into detail on how the samples were prepared but if you would like further detail, please refer to the paper (future link). We sequenced the V3/V4 region of the 16S gene, using the recommended primers and protocol (with some changes) in the workflow from Illumina.
The samples were spread across four separate MiSeq runs (2 x 300 bp) each with positive and negative sequencing controls. The data was returned in the form of a _R1.fastq.gz and _R2.fastq.gz file for each sample so these were the files I started with.
Major software used
I have used the following software in this analysis:
- FastQC v0.11.3
- USEARCH v8.1.1861. I used the 32-bit version and largely followed the UPARSE pipeline which is well-documented here but has been updated since this analysis.
- QIIME v1.9.1. I used this in a Docker environment, thanks to user kaitaolai.
- R v3.3.2 via RStudio. I used several packages which I will name as I describe them.
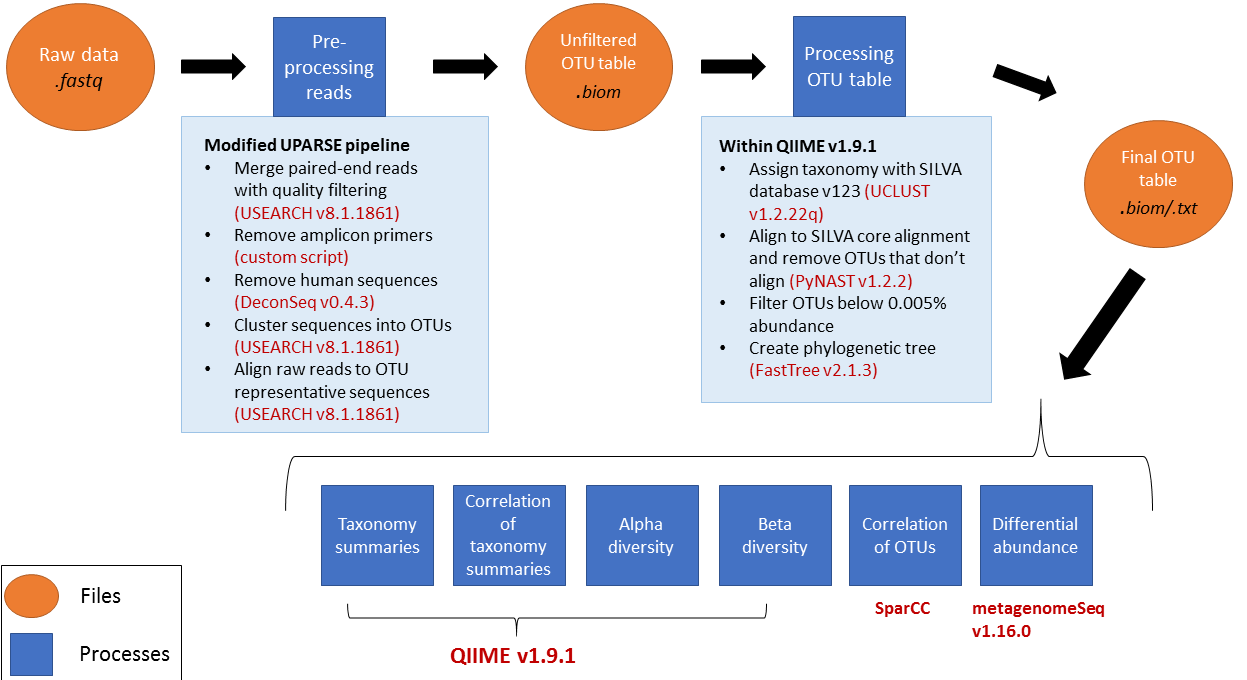
Outline of pre-processing analysis
See the diagram below for an overview of the full analysis.